

|
医疗器械警戒快讯 2025年第7期(总第221期)
发布日期:2025-08-06
医疗器械警戒快讯
(总第221期)
内容提要 加拿大Health Canada发布关于GE Healthcare公司因安全漏洞问题召回PACS系统的警示信息 美国FDA发布关于Abiomed公司血泵控制器问题的早期警示信息 美国FDA发布关于某些日本生产的奥林巴斯医疗器械的进口警示信息 美国FDA发布关于Baxter Healthcare公司因存在连接不当风险召回移动升降机组件的警示信息 美国FDA发布关于Baxter公司输液泵输注不足问题的早期警示信息 美国FDA发布关于Integra LifeSciences公司因解体风险召回Codman一次性颅骨打孔器的警示信息
加拿大Health Canada发布关于GE Healthcare公司因安全漏洞问题召回PACS系统的警示信息
发布日期:2025年7月3日 召回级别:II级 召回产品:型号为K2070NA的Centricity 制造商:GE Healthcare IITS USA Corp / GE Healthcare 召回发起日期:2025年6月20日 召回原因: GE Healthcare最近发现了一个影响AW(高端工作站)套件(医学影像存储和传输系统软件,PACS)的网络安全漏洞,该漏洞导致服务器登录凭据能够被识别,可能导致未授权的第三方登录系统修改患者数据。 (加拿大Health Canada网站)
美国FDA发布关于Abiomed公司血泵控制器问题的早期警示信息
发布日期:2025年7月 1日 警示产品:自动Impella控制器 产品用途: 自动Impella控制器是Impella导管的主要用户控制界面。Impella泵系统是提供临时性完全或部分心脏支持的血液泵,可在需要维持患者血流动力学稳定的辅助治疗期间,或需要暂时减轻心脏负荷以帮助急性病症恢复时,承担患者部分或全部血液循环功能。 警示原因: Abiomed公司声明已发现自动Impella控制器(AIC)存在可能影响检测连接Impella泵的功能问题。该泵体检测故障可能发生在所有Abiomed Impella泵型号中,且可能出现在控制台间转移或病例启动过程中。在此类情况下,AIC屏幕不会显示任何视觉警报以提示检测故障。 该问题可能导致血流动力学支持不足。心源性休克患者面临更高风险,因为长时间的支持不足可能造成患者不耐受,并导致危及生命的损伤。 截至6月13日,Abiomed尚未报告与此问题相关的严重伤害,但已有3例相关死亡病例。 采取措施: 自动 Impella 控制器 (AIC) 在连接时可能无法检测到 Impella 泵。建议配备备用AIC设备以防罕见故障发生。 6月23日,Abiomed公司向所有受影响客户发送信函,建议采取以下措施: 配备备用自动Impella控制器(AIC),以应对设备可能出现的罕见故障。 控制台间转移操作:若泵体连接到转移后的控制台,持续显示图1界面超过20秒无响应:立即将泵体切换回原控制台以维持患者支持;若原控制台显示报警信息,请更换其他可用控制台;重新连接泵体前,重启卡在图1界面的控制台。 病例启动操作:若连接泵体后,控制台持续显示图2界面超过20秒且未跳转至"检测Impella"状态:需在当前控制台重新启动病例,或更换其他控制台连接泵体。 请仔细阅读本通知并转发给院内相关人员。 若受影响产品已转至其他机构,请立即联系该机构并传达本通知内容。
(美国FDA网站)
美国FDA发布关于日本生产的某些奥林巴斯医疗器械的进口警示信息
发布日期:2025年6月24日 警示内容:美国食品和药物管理局(FDA)正在向医疗服务机构发出关于日本奥林巴斯医疗系统公司(奥林巴斯)及其子公司生产的某些医疗器械的进口警示信息。尽管奥林巴斯在解决合规问题上做出了广泛而持续的努力,但FDA仍然对其违反质量体系法规的行为表示担忧。因此,FDA已发布进口警告,以防止未来某些医疗器械进入美国,包括以下特定型号: 输尿管镜,用于开展泌尿道的各种诊断和治疗 支气管镜,用于开展呼吸道的各种诊断和治疗 腹腔镜,用于开展腹部和骨盆的各种诊断和治疗 自动内窥镜再处理器,用于对各种内窥镜进行再处理 FDA建议医疗服务机构: 一、注意FDA对奥林巴斯医疗系统公司(奥林巴斯)及其子公司在日本生产的某些医疗器械的进口警告,根据该警告,这些设备将被拒绝进入美国:进口警报89-04,原因是奥林巴斯在日本的会津生产基地未能满足质量体系相关监管要求。 二、请参阅文末链接,了解纳入进口警报中的型号和唯一标识符(UDI)。 注意进口警报不适用于与受进口警报影响的器械一起使用的相关产品(如更换部件、连接器或一次性消耗品)。 三、如果您目前正在使用受进口警告约束的器械,且器械没有遇到任何问题,则可以继续使用。 1.按照标签和再加工说明对器械(包括配件)进行清洁和再加工。 2.不要使用损坏或未通过泄漏测试的器械,因为它们可能是潜在的污染源。 3.根据制造商的说明制定例行检查和定期维护计划。 四、与你的病人讨论使用这些器械的益处和风险。FDA不建议在医疗服务机构和患者没有讨论获益和风险的情况下取消或延迟手术。 五、及时完成不良事件的报告,以帮助我们识别和更好地了解与这些器械相关的风险。 警示原因 FDA确定某些工厂不符合现行良好生产规范(CGMP)和包括最严重类型的召回在内的各种报告要求,发布了警告信和进口警报。 采取措施 FDA已经对奥林巴斯的质量体系要求和合规问题采取了几项行动。 FDA正在继续与奥林巴斯合作,以加快与正在进行的违规行为相关的纠正措施,将患者的风险降至最低,并可能酌情采取进一步行动。FDA在确保患者医疗器械安全有效方面发挥着重要作用。 如果有新的或额外的信息,FDA将继续通知医疗服务机构和公众。 受进口警报影响的奥林巴斯医疗器械清单,详细信息点击链接获取:https://www.fda.gov/medical-devices/letters-health-care-providers/import-alerts-certain-olympus-medical-devices-manufactured-japan-letter-health-care-providers。 (美国FDA网站)
美国FDA发布关于Baxter Healthcare公司因存在连接不当风险召回移动升降机组件的警示信息
发布日期:2025年7月23日 召回级别:此次召回涉及从使用或销售地点移除器械。美国食品药品监督管理局(FDA)已将此次召回确定为最严重类型。若您继续使用该器械,可能会导致严重伤害或死亡。 召回产品: Q-Link 13,产品编号3156509 该产品与以下移动升降机配合使用: Uno 102 EE移动升降机;产品编号:2010004 Viking L移动升降机;产品编号:2040044 Viking XL移动升降机;产品编号:2040043 Viking M移动升降机;产品编号:2040045A Viking S移动升降机;产品编号:2040006 Viking XS移动升降机;产品编号:2040007 LikoLight移动升降机;产品编号:2030001
LikoScale适配器套件,产品编号3156232 该产品需与以下LikoScale适配器套件配合使用 LikoScale 200配件;产品编号3156225 LikoScale 350配件;产品编号3156228 LikoScale 400配件;产品编号3.156226 产品用途: Q-Link 13是一个可选的升降组件,当连接到用于吊带杆的快速释放挂钩时,可与移动式升降机结合使用。LikoScale适配器套件包含Q-Link 13组件,并存在连接不当的风险。移动式升降机适用于最常见的升降场合,如将患者从床上转移到轮椅上,往返于厕所和浴缸之间,以及从地面升降。部分移动式升降机也可用于步态训练。 召回原因: Baxter公司表示,Q-link 13可能会使吊带杆和其他配件上使用的快速释放钩发生不当连接(误锁)。误锁的部件最初可能会承受重量,但可能会从Q-link上松动,导致脱落和掉落。这可能会导致患者摔倒而造成严重伤害。护理人员试图阻止患者摔倒时也可能面临受伤风险。 Baxter公司已报告三例与此问题相关的严重伤害事件和一例死亡事件。 召回措施: Baxter发出了两封单独的紧急产品召回函,通知受影响产品的收货人。 第一封日期为2025年5月30日的信件被发送给医疗机构,收件人为生物医学工程、护理总监或分销商/经销商。 第二封信日期为2025年6月2日,收件人为家庭患者。 这两封信都包括以下建议行动: 1.立即定位并停止使用与受影响产品表中列出的产品一起使用的所有Q-link 13组件。 2.Baxter将提供更新的Q-link Mobile版本的替代Q-link 13产品,旨在提高可用性并降低患者和护理人员的潜在风险。一旦有足够的Q-link Mobile组件可用,将发送后续通信,包括请求更换的说明。 3.将此信张贴在受影响的移动电梯存放或使用区域。 4.如果您是通过分销商或批发商购买的,请注意,通过Baxter客户门户网站的回复不适用。相反,如果需要回复,请遵循分销商或批发商的具体说明。 (美国FDA网站)
美国FDA发布关于Baxter公司输液泵输注不足问题的早期警示信息
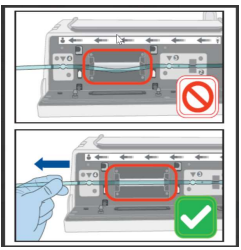
发布日期:2025年7月22日 警示产品:Novum IQ大容量泵 产品用途:Baxter Novum IQ大容量泵旨在在医生或其他认证医疗保健专业人员的指导或监督下,为患者提供静脉输液、血液和血液制品。 警示原因: Baxter公司表示,Novum IQ 大容量输注泵(LVP)在输注速率由一个流速切换到更高流速(例如变更速率或推注)时,存在输注不足的潜在风险。具体而言,当第二个流速超过第一个流速的两倍时,风险就会发生。输注不足的程度不一,取决于较低流速的输注速率、该速率下运行的持续时间以及速率变化的幅度。泵在低速率下运行时间越长,速率变化幅度越大,输注不足的情况就越严重。在最严重的情况下,可能完全不输注。 此外,Baxter公司还发现,客户关于过量输注或输注不足的报告有所增加,可能是由于输注管组装不当造成的。如果输注管未正确装入泵通道,可能会导致泵的实际输注速率高于或低于设定速率。 输注不足、过量输注或未输注所需溶液或药物可能导致的有害后果因患者个体、所治疗的疾病状况以及所输注的液体、药物或其他治疗方式的不同而有所不同,可能从轻微或短暂的不良事件到严重伤害不等。高风险及脆弱患者可能会遭遇严重的健康后果,包括血流动力学不稳定、心律失常、镇静不足、高血糖和血栓栓塞事件等。 截至6月27日,Baxter公司已报告与该问题相关的79例严重伤害事件和2例死亡事件。 采取措施: 在首次具备安全条件的情况下,更换输液管路组件,或更换为配有新输液管路的另一台输注泵。 如果更换输注泵和/或输液管路所需的时间延误不可接受,用户在启动推注输注或进行超过 100% 的速率变更前,应将输液管路组件向下游(靠近患者方向)移动约 0.5 英寸。 2025年7月14日,Baxter公司向所有受影响客户发送了一封信函,建议采取以下措施: 1.请参阅 Novum IQ LVP 操作手册第 4.4 节《卸下输液管路组件》的说明,正确卸下输液管路组件。 2.为防止自由流动,请确保下游滚轮夹完全关闭。 3.卸下输液管后,将滑夹向容器方向移动 0.5 英寸,以实现输液管下移。 4.移动滑夹后,请根据 Novum IQ LVP 操作手册第 4.3 节《装载输液管路组件》的说明重新装载输液管。 5.输液管装入后,关闭泵门并弹出滑夹,确保下游滚轮夹完全打开。 6.在具备安全条件时,尽早更换输液管路,或定期检查输注是否以设定速率进行。 装载输液管路组件前,确保泵门完全打开。 如下图所示,管在泵送通道中绷紧并加载,没有松弛。
即使您没有剩余库存,也要按照随附的回复说明表上的说明确认收到此通知。 请将此通信的副本转发给首席医疗官、医疗主任、药房主任、机构风险管理负责人、采购/中央供应主任、麻醉主任以及贵机构内使用受影响产品的任何其他部门。 (美国FDA网站)
美国FDA发布关于Integra LifeSciences公司因解体风险召回Codman 一次性颅骨打孔器的警示信息
发布日期:2025年7月16日 召回级别:此次召回涉及从医疗器械使用单位或销售机构召回部分器械。美国食品药品监督管理局(FDA)已将此次召回识别为最严重的类型。如果继续使用该器械,可能会导致严重伤害或死亡。 召回器械:Codman一次性打孔器;Codman开颅手术套件 UDI及批号参见FDA网站: https://www.fda.gov/medical-devices/medical-device-recalls/cranial-drill-recall-integra-lifesciences-recalls-codman-disposable-perforators-due-risk-device 召回原因:Integra LifeSciences正在召回部分Codman一次性颅骨打孔器和开颅手术套件。Codman一次性颅骨打孔器是神经外科专用手术工具,用于颅骨钻孔开窗,其设计特点是在钻孔完成后自动停转。本次召回是原因是该器械外套管存在超声波焊接缺陷,该缺陷可能会导致器械在开颅手术前、中、后期发生解体。部分产品可能无法立即停转。使用有缺陷的产品可能会导致严重伤害,包括:硬脑膜损伤、脑部出血、脑组织损伤;出现手术时间延长、不可逆脑损伤后果,甚至导致死亡。目前已经收到10份伤害报告,涉及手术延误、术中器械卡入颅骨、器械碎片取出困难、出血、硬脑膜损伤和脑损伤。目前还没有收到死亡报告。 召回措施:Integra LifeSciences已向所有受影响的客户发送了一封医疗器械紧急召回信,建议立即停止使用并隔离、申请退回相关产品。 (美国FDA网站)
|



|