

|
医疗器械警戒快讯 2025年第12期(总第226期)
发布日期:2026-01-04
医疗器械警戒快讯
(总第226期) 内容提要
澳大利亚TGA发布关于Olympus公司因刀头断裂风险召回一次性使用黏膜切开刀的警示信息 发布日期:2025年12月16日 召回级别:I级 召回产品:KD-640L及KD-645型一次性使用黏膜切开刀 召回原因:Olympus公司在收到关于KD-640L和KD-645L一次性使用黏膜切开刀在使用过程中三角形刀头断裂的投诉后进行了调查,确定切开刀因过热和烧焦而劣化,可能导致使用过程中刀头断裂。 根据使用说明书中的警告,用户应间歇性激活切开刀输出,如果设备的三角形刀头变红,应立即停止激活输出。 未按照使用说明书提示使用这些设备可能会导致患者受伤,例如产品刀头/碎片脱落到患者体内。这可能会导致手术时间延长,从而增加麻醉时间,并可能需要额外的影像检查或手术来取出异物。其他可能的危害包括烧伤、穿孔,如果刀头无法在患者体内找到,则可能出现异物反应,以及可能的吸入风险,尤其是在喉咽部位进行手术时。 采取措施:用户应阅读客户信函,以确保了解使用说明书和标签的更新,其中包含新的图表和以下警告内容: 当三角形刀头未接触组织、组织已碳化或碳化组织附着在刀头或切开刀上时,切勿激活高频电流。否则,过大的电流放电可能会对非目标组织造成意外伤害。如果三角形刀头因持续过度放电而变红,请立即关闭电源,以防止对组织造成热损伤。如果此时将切开刀从外鞘中拔出,三角形刀头可能会掉落。 客户还应按照现有的使用说明书警告,正确激活KD-640L和KD-645L电刀。 (澳大利亚TGA网站)
澳大利亚TGA发布关于Emergo公司因螺丝断裂风险召回HFD100头部固定装置的扭矩螺丝组件的警示信息 发布日期:2025年12月15日 召回级别:Ⅱ级 涉及器械:HFD100头部固定装置的扭矩螺丝组件 零件编号、UDI编号等参见TGA网站: https://apps.tga.gov.au/PROD/DRAC/arn-detail.aspx?k=RC-2025-RN-01053-1 召回原因:HFD100头部固定装置的扭矩螺丝组件与颅骨夹组件相连接,能够为头部固定装置提供三个固定点之一的钉式固定点。据报告,扭矩螺丝的外壳可能会出现裂纹或发生径向分离,这种情况会在临床使用期间或使用前即观察到。如果使用了有裂纹或损坏的扭矩螺丝,可能导致术中患者头部稳定性不足、手术精度下降、手术延误、需要临时替换头部固定方法,增加患者受伤害的风险。 截至目前,已收到3起投诉,反映在将扭矩螺丝推进至HFD100头部固定装置的过程中,螺丝出现裂纹或断裂的情况。尚未收到与此问题相关的伤害报告。 采取措施:Emergo公司将对HFD100头部固定装置的扭矩螺丝组件改进设计并更新替换。更新后的组件计划于2026年1月开始上市,Emergo公司将联系相关用户进行更换。在更换之前,用户应采取如下控制措施: (澳大利亚TGA网站)
美国FDA发布关于GE HealthCare公司麻醉系统的风险警示信息
受影响的产品:FDA注意到,GE HealthCare公司已致信受影响的客户,建议更新某些Carestation 600和700系列麻醉系统的使用说明:
除上述产品外,受影响的设备还可以与以下产品一起作为备件安装:
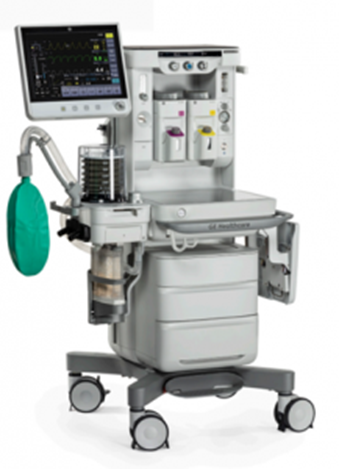
产品用途:Carestation麻醉系统旨在为包括新生儿、儿童和成人在内的广泛患者提供全身吸入麻醉和通气支持。 采取措施:在使用受影响的设备时,请始终确保设备与交流电源的安全连接。如果系统失去交流电源导致意外关闭,请遵循以下说明。 2025年11月14日,GE HealthCare公司向所有受影响的用户发送了一封信,建议采取以下行动: 始终确保设备与交流电源有安全连接。 FDA目前正在审查有关这一潜在高风险设备问题的信息,并将在获得重要新信息时向公众通报。 发布警示信息的原因:GE HealthCare公司已经意识到,如果拔掉交流电源或发生交流电源故障,包含某些电源管理板的Carestation 600和700系列麻醉系统可能会意外关闭。 麻醉系统只有在极少数情况下使用电池供电,即交流电源丢失,并且没有连续的备用应急电源。如果交流电源中断,Carestation 600和700系列麻醉系统将不会自动切换到电池供电模式,并在电源恢复后重新启动。如果出现此问题,可能会导致机械通气、人工通气和挥发剂输送暂时中断。重启后,系统不会恢复到之前的通气设置。 如果这种情况没有被用户识别和处理,失去通气可能会危及生命。 截至2025年11月26日,GE HealthCare公司尚未报告与此问题相关的任何严重伤害或死亡。 (美国FDA网站)
美国FDA发布关于Abbott Diabetes Care公司血糖监测传感器的风险警示信息 发布日期:2025年12月2日 受影响的产品:FDA注意到,Abbott Diabetes Care公司已向经销商、医疗保健提供者和受影响的客户发出信函,建议从用户或销售机构移除某些血糖监测传感器: 受影响批次的完整列表详见: https://www.fda.gov/media/189900/download?attachment FreeStyle Libre 3阅读器和移动应用软件不受影响。此外,没有其他Libre产品(FreeStyle Libre 14 day、FreeStyle Libre 2、FreeStyle Libre 2 Plus或Libre Pro传感器)或Abbott生物可穿戴设备受到影响。 产品用途:FreeStyle Libre 3和FreeStyle Libre 3 Plus连续血糖监测系统是实时连续血糖监测(CGM)设备,具有报警功能,适用于4岁及以上人群的糖尿病管理。除非另有说明,这些设备旨在取代血糖测试,用于糖尿病治疗决策。 该系统还可监测趋势,具有跟踪模式,帮助检测高血糖症和低血糖症的发作,从而促进急性和长期治疗调整。系统读数的解释应基于血糖趋势和一段时间内的几个连续读数 该系统还旨在与数字连接设备自主通信。该系统可以单独使用或与这些数字连接设备结合使用,使用户可以手动控制治疗决策。 召回原因:Abbott Diabetes Care公司表示,某些FreeStyle Libre 3和FreeStyle Libre 3 Plus传感器提供不正确的低血糖读数。如果未被检测到,长期不正确的低血糖读数可能会导致糖尿病患者做出错误的治疗决定,例如过量摄入碳水化合物或跳过或延迟摄入胰岛素。这可能会带来严重的健康风险,包括潜在的伤害或死亡,或其他不太严重的并发症。 截至2025年11月14日,Abbott公司报告了736起严重伤害,以及7起与此问题相关的死亡。 采取措施: 患者应验证其传感器是否受到影响,并立即停止使用和处理受影响的传感器。 2025年11月24日,Abbott Diabetes Care公司向所有受影响的客户发送了一封信函,建议采取以下措施: FDA目前正在审查这一潜在的高风险设备问题,并将在有新的重要信息时通知公众。 (美国FDA网站)
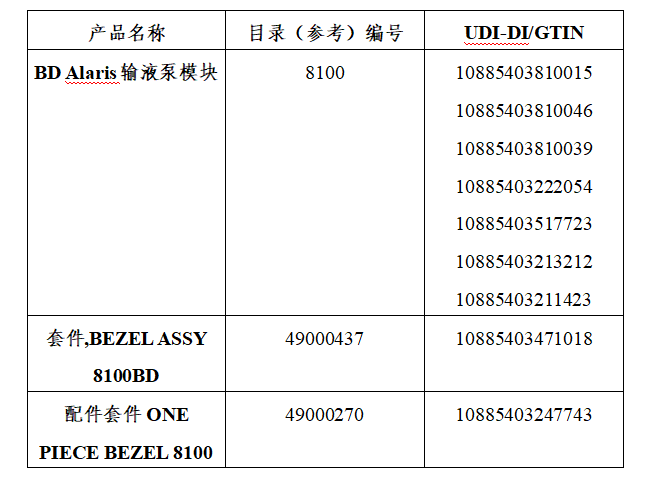
美国FDA发布关于Becton Dickinson公司因BD Alaris输液泵问题更新产品使用说明的警示信息 发布日期:2025年11月28日 此次召回涉及更新器械的使用说明,而非从使用单位或销售机构移除器械。FDA将此次召回认定为最严重的类型。如果您不按照更新后的说明继续使用,可能会造成严重伤害甚至死亡。 受影响的产品: FDA获知CareFusion 303旗下子公司Becton Dickinson(BD)已向受影响客户发函,建议用户对掉落或剧烈震动过的Alaris输液泵模块采取额外预防措施。 产品用途:BD Alaris系统是一种模块化输液泵和监测系统,通过临床认可的静脉注射(IV)、动脉内注射(IA)、皮下注射、硬膜外注射或液体冲洗等方式,为成人、儿童和新生儿患者提供连续或间歇性输液。 召回原因:BD公司表示,Alaris输液泵模块掉落后可能会损坏内部组件,这些组件可能不易察觉或难以察觉其存在问题。更具体地说,掉落或剧烈震动可能损坏为关键泵组件提供机械基础的Alaris输液泵模块挡板配件。这种损坏可能导致输注不足、输注过量、流量失控或Alaris输液泵模块无法校准。 泵送装置受损可能阻止设备无法通过泵送循环准确输送流体,导致输注过量或不足。输注过量和不足可能导致包括严重伤害和死亡在内的严重不良健康后果,具体取决于药物类型(及其临床效果)、患者危急程度以及流量不准确的程度。 截至9月22日,BD公司报告了2起严重伤害,暂无与此事相关的死亡事件。 采取措施:不要使用曾经掉落或剧烈震动过的设备。应立即移除曾经掉落或剧烈震动过的设备,并由合格的服务人员按照使用说明进行彻底测试和检查后再使用。 10月17日,BD公司向所有受影响客户发送了一封信,建议采取以下措施: (美国FDA网站)
|



|