

|
医疗器械警戒快讯 2026年第3期(总第229期)
发布日期:2026-05-09
医疗器械警戒快讯
(总第229期)
内容提要
美国FDA发布关于Philips公司召回Trilogy Evo系列呼吸机的警示信息 发布日期:2026年4月15日 召回级别:此次召回涉及对产品进行纠正,不涉及将其从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果在未进行纠正的情况下继续使用该产品,可能会导致严重伤害或死亡。 受影响的产品 FDA获悉,Philips公司已向受影响的客户发出通知,建议在继续使用前对所有Trilogy呼吸机进行。
受影响产品完整列表详见FDA网站: https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/ventilator-correction-philips-issues-correction-trilogy-evo-platform-ventilators 产品用途 Philips Respironics Trilogy Evo、Evo O2、EV300和Evo Universal呼吸机为需要机械通气的患者提供持续气道正压通气(CPAP)和间歇气道正压通气支持。这些产品适用于体重至少2.5kg的儿童至成人患者,可在医院和卫生保健机构中使用,也可用于患者的非紧急转运。Trilogy EV300和Trilogy Evo也可供需要居家机械通气的患者使用。 召回原因 Philips公司已发现三个影响Trilogy Evo系列呼吸机的问题: 非气动式雾化器使用风险。在该系列呼吸机上使用非气动式雾化器(如振动网式)会改变气流特性,导致呼吸机泄漏量估算计算出现偏差,造成设定通气量与患者实际接收通气量不符,引发通气不足、血氧饱和度降低、治疗不当等呼吸系统问题。 根据雾化器在患者回路中的放置位置,气溶胶可能会进入呼吸机并在内部流量传感器上积聚。随着时间的推移,这种堆积可能会干扰传感器准确测量气流的能力,导致气流计算错误,并可能影响给予患者的治疗。在这种情况下,应更换内部流量传感器。 气溶胶可能进入呼吸机并在内部流量传感器上积聚,影响气流测量准确性,进而导致潮气量过量输送、FiO2输送不足,极端情况下可能引发通气失效,造成肺过度充气、呼吸不适等危害。 堵塞报警时序异常,在某些情况下,阻塞报警不会在相关标准规定的时间范围内(两个呼吸周期或5秒)触发;在部分通气模式下,无论是否启用备用通气频率,报警可能延迟长达四个呼吸周期。 截至2026年3月6日,Philips公司已报告3份与这些问题相关的严重伤害事件,未出现死亡报告。 召回措施 将所有Trilogy Evo系列呼吸机的软件版本更新至1.05.15.00。停止在所有Trilogy Evo系列呼吸机上使用非气动式雾化器。查阅最新的用户手册附录。 2026年3月2日,Philips公司向所有受影响的客户发送了信函,建议采取以下措施:
(美国FDA网站)
美国FDA发布关于Draeger公司因通气故障问题召回Atlan麻醉工作站的警示信息 发布日期:2026年4月14日 召回级别:此次召回涉及对产品进行纠正,不涉及将其从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果在未进行纠正的情况下继续使用该产品,可能会导致严重伤害或死亡。 召回产品
产品用途 Atlan是一款麻醉工作站,适用于成人、儿童及新生儿的麻醉。该产品可用于机械通气、手动通气、压力支持下的自主呼吸及自主呼吸。根据手术或诊断干预期间的预期用途,该产品适用于吸入式麻醉和/或患者通气。 召回原因 Draeger公司收到报告称,Atlan系列麻醉工作站在使用前显示活塞通风故障,且在使用过程中发生机械通气故障。Draeger公司经过调查后认为上述错误是由于制造过程中的某个时间段引入了杂质导致的。截至2025年9月9日,Draeger公司尚未报告任何与该问题相关的严重伤害或死亡事件。 召回措施 对所有受影响产品进行通风电机组件检查,必要时予以更换。2025年9月9日,Draeger公司向客户发送了紧急医疗器械纠正通知,并就产品发生错误时的纠正方法作出如下指导:
(美国FDA网站)
美国FDA发布关于Merit Medical公司因设计缺陷召回透析导管的警示信息 发布时间:2026年4月14日 召回级别:本次召回涉及将特定器械从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果继续使用该产品,可能会导致严重伤害或死亡。 召回产品 FDA获悉,Merit Medical公司已向受影响的客户发出通知函,建议将特定的透析导管从使用单位或销售机构移除。此导引器是以下套件的组件:
完整产品清单详见FDA网站: https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/dialysis-catheter-recall-merit-medical-removes-16f-dual-valved-splittable-sheath-introducer 产品用途 16F双阀可撕开导管鞘用于多种Merit成品器械的组件,并封装在成品器械的无菌屏障内。该导引器用于将导管导入至血管系统。 召回原因 Merit Medical公司正在召回16F双阀可撕开导管鞘,原因是存在设计缺陷,可能导致导引鞘无法按预期撕开。该导引鞘被用于多种Merit的成品器械中。导引鞘未能按预期撕开,可能导致出血、异物残留、手术延误、栓塞/血栓形成、导管功能受损以及丧失未来血管通路。截至2月23日,Merit Medical公司已报告2份与该问题相关的严重伤害事件,无死亡事件。 召回措施 请勿使用Merit Medical公司的16F双阀可撕开导管鞘产品。2月5日,Merit Medical公司向所有受影响的客户发送了通知;2月27日,发送了补充通知;3月23日,向所有受影响的客户发送了修订通知。这些通知建议采取以下措施:
(美国FDA网站)
美国FDA发布关于Abiomed公司召回Impella心室辅助系统冲洗套件的警示信息 发布日期:2026年4月3日 召回级别:此次召回所涉及的产品以及对相关产品的处理建议均未发生变化。本次召回涉及将特定器械从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果继续使用该产品,可能会导致严重伤害或死亡。 受影响的产品 FDA获悉,Abiomed公司已向受影响客户发出信函,建议将某Impella冲洗盒和Impella RP泵组从使用单位或销售机构移除。
产品用途 冲洗套件将冲洗液输送至Impella导管。冲洗液从冲洗套件流经导管到达微型轴流血泵,以防止血液进入电机。 召回原因 Abiomed公司报告称,第一代冲洗套件冲洗泄漏的风险在增加。因此,当更新的第二代冲洗套件推向市场后,Abiomed公司将召回第一代冲洗套件。 如果发生冲洗套件泄漏,用户将在自动化Impella控制器(AIC)上看到“Purge Pressure Low(冲洗压力低)”警报;请参见下文警报示例: 若未及时处理冲洗泄漏,可能导致冲洗压力降低。这可能引发生物材料渗入,进而导致泵意外停机。泵停机可能导致血流动力学支持丧失,从而危及患者生命。 截至2月3日,Abiomed公司已报告4份与该问题相关的严重伤害事件,无死亡报告。 采取措施 识别并停用所有第一代冲洗套件。若无第二代冲洗套件可用且必须使用第一代冲洗套件,则需加强对冲洗系统的监测,若触发“Purge Pressure Low(冲洗压力低)”警报,请参考使用说明书(IFU)。 2026年2月18日,Abiomed公司向所有受影响的客户发出了一封信,建议采取以下行动:
(美国FDA网站)
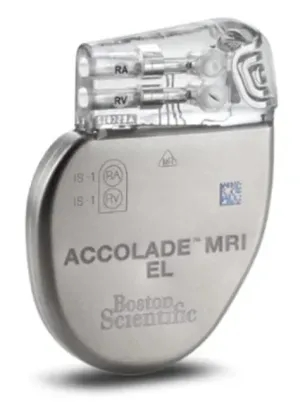
澳大利亚TGA发布关于Boston Scientific公司起搏器和心脏再同步化治疗起搏器的警示信息 发布时间:2026年4月1日 该市场行动已更新为包括有关软件纠正、医务人员行动等涉及所有CRT-P和双室延长寿命(DR-EL)产品的新信息。这些更新是在2025年9月和2024年12月发布的网络声明基础上发布的。 Boston Scientific公司正在对ACCOLADE和VISIONIST起搏器进行产品校正和产品警报。正如之前在2024年12月公布的那样,Boston Scientific公司建议,ACCOLADE单室(SR)、双室(DR)标准寿命(SL)和DR延长寿命(EL)起搏器的一些产品和VISIONIST心脏再同步化治疗起搏器(CRT-Ps)永久进入功能有限的安全模式的风险增加。Boston Scientific公司正在通知医疗保健提供者和患者可以获得软件补丁。此软件补丁可以降低产品进入安全模式的风险,并纠正以前的软件补丁可能出现的意外问题。 受影响的产品
更新内容 软件更新可用于降低2025年9月发布软件补丁纠正的产品在医疗机构之外在正常、高功率操作期间永久进入安全模式的风险。在一些患者中,产品永久进入安全模式与起搏器不能充分调节心律和心率的风险有关。除了先前描述的意外行为外,Boston Scientific公司还确定了3869 v2.04型的第三种意外问题。在特定情况下,确定电池状态和剩余使用时间的电池测试可能冻结,直至再次检查。因为一些产品可能尚未达到预期寿命,市场行动已扩大到涵盖所有ACCOLADE CRT-P和双室延长寿命(DR-EL)产品,不管其使用期限如何。 警示原因 与此问题发生相关的风险包括:
采取措施(患者)
采取措施(医护人员)
详细信息可以在提供给植入该产品的临床医生的客户信中找到。 (澳大利亚TGA网站)
|




|