

|
医疗器械警戒快讯 2026年第5期(总第231期)
发布日期:2026-06-04
医疗器械警戒快讯
(总第231期)
内容提要 美国FDA发布Abiomed公司心脏泵控制器问题的早期警示信息 美国FDA发布关于React Health公司因氧气泄露风险召回VOCSN V+Pro呼吸机的警示信息 美国FDA发布关于Omnicell公司因贴标错误风险召回自动化配药系统配合使用的特定注射器标签的警示信息 美国FDA发布关于Bolton Medical公司因释放困难问题召回Relay Pro胸主动脉覆膜支架的警示信息 美国FDA发布关于Draeger公司麻醉工作站制造错误问题的早期警示信息 美国FDA发布关于Arrow International公司召回含有Merit Medical可撕开导管鞘透析导管套件的警示信息
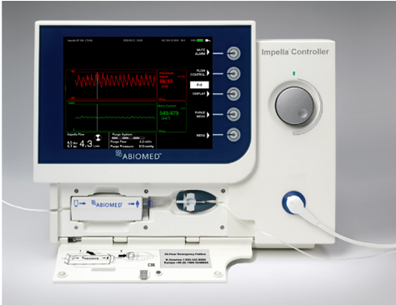
美国FDA发布Abiomed心脏泵控制器问题的早期警示信息
发布日期:2026年5月21日 警示器械:
FDA发现Abiomed已向受影响的客户发布紧急医疗器械召回(更正)通知,建议更新所有自动化心室辅助装置控制器(AIC)的使用说明。受影响的设备:
完整产品清单见原始网页:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/early-alert-heart-pump-controller-issue-abiomed。
器械用途:AIC是驱动装置的基础用户控制界面。它可以控制驱动装置并监测相关警报。驱动疗法旨在减少心脏的工作,为循环系统提供支持,使心脏有时间恢复。 警示原因: Abiomed已经确定,当患者接受左心室辅助装置治疗并经历较长时间(约80分钟)无残余脉搏(主动脉放置信号<12 mmHg)时,由于内部软件错误,自动化心室辅助装置控制器(AIC)可能会被迫重新启动。如果左室压(LVP)计算处于活动状态(支持水平高于P-3)时,左心室(LV)压力突然发生变化,则可能发生这种情况。禁用主动脉位置信号和LVP的显示并不会阻止AIC重新启动。 在重新启动过程中,AIC屏幕将在没有进一步警报的情况下变黑,根据目前的数据,泵将停止大约35秒,在此期间患者没有驱动系统的支持,可能会发生反流。AIC重启后,泵会自动恢复到之前的P水平。用另一个AIC替换AIC不会解决AIC重新启动的可能性。 由于在系统重新启动时缺乏血流动力学支持,没有替代机械支持的患者可能会增加发生严重伤害或死亡的风险。 产品没有被移除,医院库存可以继续使用。为了解决这个问题,Abiomed正在对AIC进行软件更新。 截至4月27日,Abiomed报告了与此问题相关的两例严重伤害和一例死亡报告。 采取措施: 关注当与左心室心脏辅助装置一起使用时,软件错误可能导致AIC重新启动。 5月14日,Abiomed向所有受影响的客户发送了一封信,建议采取以下措施: 跟进FDA网页了解最新情况。FDA目前正在审查有关这一潜在高风险设备问题的信息,并将在获得重要新信息时向公众通报。 (美国FDA网站)
美国FDA发布关于React Health公司因氧气泄露风险召回VOCSN V+Pro呼吸机的警示信息
发布日期:2026年5月21日 本次召回涉及将特定器械从使用或销售地点撤下。FDA已确定本次召回属于最严重类型。若继续使用,该器械可能导致严重伤害或死亡。 召回产品:见下图
FDA获悉,Ventec(以React Health名义开展业务)已向受影响的客户发出通知函,建议将特定的VOCSN V+Pro设备从使用或销售地点撤下。受影响器械完整清单请见FDA网站:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/ventilator-recall-react-health-removes-vocsn-vpro-ventilators 器械用途:VOCSN一体化呼吸系统(VOCSN Unified Respiratory System)旨在为需接受机械通气的患者提供持续或间歇的呼吸机支持,适用于有创及无创通气场景。VOCSN V+Pro设备仅提供 V(通气)功能模块,并具备外部高压及低压氧气接入能力。 召回原因: React Health正在撤下受影响设备,原因是制造工艺出现偏差,可能导致难以察觉的氧气泄漏状况。 因生产测试配置错误,部分设备在生产测试阶段可能未覆盖全部预期高压工况的评估。因此,可能发生氧气泄漏,导致在通气前或通气期间输送的吸入氧浓度(FiO2)处于规范限值之外。 若发生氧气泄漏,实际输送的吸入氧浓度可能低于设定水平。在富氧环境中,泄漏还可能增加火灾风险。上述情况可能导致患者严重受伤或死亡。 截至3月4日,React Health尚未报告任何与该问题相关的严重伤害或死亡事件。 召回措施: 识别并停止使用受影响设备。请使用非受影响呼吸机为患者提供支持。 3月23日,React Health向所有受影响的客户发送通知函,建议采取以下行动: (美国FDA网站)
美国FDA发布关于Omnicell公司因贴标错误风险召回自动化配药系统配合使用的特定注射器标签的警示信息
发布日期:2026年5月21日 此次召回包括将特定器械从使用或销售的地方移除。FDA已将此次召回确定为最严重的类型。如果您继续使用该器械而不进行纠正,可能会导致严重伤害或死亡。 召回产品: FDA获悉,Omnicell公司已向受影响的客户发出信函,建议将与 i.v.STATION 自动化配药系统配合使用的特定注射器标签从其使用或销售地点移除: IVSTATION 注射器无菌标签(40MM×40MM)- 部件号 258920028 IVSTATION 注射器无菌标签(20MM×40MM)- 部件号 258920029 这些标签各自独立包装在塑料外包内,背衬为白色。 器械用途: i.v.STATION 是一种自动化设备,可限制在静脉用药的制备和配药过程中手动处理西林瓶、溶液、静脉输液袋和注射器的需求,同时将药物间的交叉污染降至最低。作为该设备自动化功能的一部分,它会为配制的制剂贴上标签,以便进行识别和追溯。 召回原因: Omnicell 收到一份可能影响使用 i.v.STATION 生产的无菌注射器标签的贴标错误报告。Omnicell 发现,在使用受此次召回影响的无菌标签时,i.v.STATION 各打印机之间存在标签检测行为不一致的情况。在使用上述受影响的标签时,标签检测不一致可能导致配药完成后出现无标签或错误标签的注射器制剂。 截至2026年4月10日,Omnicell 报告称尚无与该问题相关的严重伤害或死亡事件。 采取措施: 2026年4月10日,Omnicell 向所有受影响的客户发出信函,建议采取以下措施: 请勿使用受此次召回影响的标签。如果您或您的员工发现任何受影响的标签,请立即隔离并联系 Omnicell 技术支持部门。 恢复使用之前订购的背衬为黄色的非无菌注射器标签(部件号 HR060027 和 HR060071)。 按照您现有的复核流程,采取审慎的药师核查措施,以验证标签的准确性。 将此信息分享给您组织内所有需要知悉的人员。 (美国FDA网站)
美国FDA发布关于Bolton Medical公司因释放困难问题召回Relay Pro胸主动脉覆膜支架的警示信息
发布日期:2026年5月20日 FDA 发布本次警示信息,向公众通报高风险医疗器械问题。FDA已将此次召回定性为最严重召回类型。若继续使用该设备,可能会导致严重伤害或死亡。 召回产品: 器械名称:Relay Pro 胸主动脉覆膜支架系统 型号规格:N4非裸支架配置,32mm及以上规格 器械用途:Relay Pro 胸主动脉覆膜支架系统是一种微创医疗设备,通过放置覆膜支架修复胸主动脉受损或变薄段落,包括动脉瘤、夹层或破裂等病变部位,以此增强血管强度并维持正常血流。 召回原因: Bolton Medical 说明,在某些情况下,由于近端锁扣与外控制管断开,导致植入物无法从输送系统解脱。使用者可能会发现,在向后滑动顶端固定器时无阻力感,并伴有近端支架无法释放的情况。在手术过程中的这一阶段,植入物无法被重新回收。这种失效模式直到术中发生时才能被识别。 释放覆膜支架困难可能导致手术延迟、覆膜支架移位以及无法释放覆膜支架。可能需中转开放手术修复卡扣,存在致死风险。此类故障可无征兆突发,且暂无针对性设备应急处理方案。 截至4月23日,Bolton Medical 已报告3例与该问题相关的死亡报告,包括1例主动脉穿孔,以及2例转为开放手术后因卒中导致患者死亡。 召回措施: 在使用受影响的 Relay Pro 器械前,考虑替代覆膜支架选项。在使用受影响器械前,请仔细阅读《使用说明书》中包含的现有解救方法。 4月23日,Bolton Medical 向所有受影响客户发送了一封信函,建议采取以下措施: 在缓解措施到位之前,考虑在使用受影响的 Relay Pro 器械前,优先使用替代覆膜支架方案。 术中若出现难以释放近端支架、向后滑动顶端固定器无阻力的情况,优先参照《使用说明》中现有应急方法处理;若问题仍未解决,需依据临床判断及时决策,必要时转为开放手术。 通知所有Relay Pro设备相关使用人员,告知支架移植物无法从输送系统释放的全新处理指南;若相关设备已流转至其他医疗机构,需转交本通知副本并指导其落实相关措施。 将《医疗器械紧急召回(更正)通知》副本与设备《使用说明书》一并存档保存,并在医疗器械存放处张贴通知副本。 (美国FDA网站)
美国FDA发布关于Draeger公司麻醉工作站制造错误问题的早期警示信息
发布日期:2026年5月15日 CDRH发布此早期警示信息,旨在向公众通报一个可能存在高风险的器械问题。美国食品药品监督管理局(FDA)将随时向公众通报最新情况,并在获得重要新信息时更新此网页。 警示器械: FDA获悉Draeger公司已向受影响的客户发出信函,建议将某些Atlan A350、Atlan 350XL 麻醉工作站从使用或销售地点撤下。 受影响的器械: 器械名称:麻醉工作站 型号:Atlan A350、Atlan 350XL UDI:Atlan A350:04048675556176 / 8621500 Atlan A350 XL:04048675556183 / 8621600 受影响产品完整列表详见FDA网站:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/early-alert-anesthesia-machine-issue-draeger-inc 器械用途:Atlan A350 和 A350 XL 麻醉工作站适用于成人、儿童和新生儿的麻醉。它们可用于:自动通气/手动通气/压力支持下的自主呼吸/自主呼吸。该设备用于在外科手术或诊断操作期间实施吸入麻醉。 召回原因: Draeger公司正在扩大其2024年10月纠正行动的范围,将更多批次的Atlan A350、Atlan A350XL麻醉工作站纳入纠正。这次纠正行动原因为某些批次的麻醉工作站因制造错误可能造成活塞呼吸机在使用前就失效,或是在使用过程中发生机械通气故障。 如果错误在使用前发生(例如待机状态或系统测试阶段),将无法启动机械通气;如果错误在手术过程中发生,机械通气可能中断,设备会通过显示“呼吸机错误!!! ”提示使用者。上述两种情况下,手动通气或自主呼吸功能仍可正常使用,此时必须为患者进行手动通气,避免造成伤害。即使通气故障警报持续触发,Atlan A350、Atlan A350XL麻醉工作站仍可无限制提供手动/自主通气模式、新鲜气体与麻醉药剂输送,以及所有监测功能。 Draeger公司将为所有涉事设备免费更换呼吸机电机组件,当地Draeger公司服务代表将联系客户,安排更换时间。 使用涉事产品可能造成严重健康后果,包括缺氧(低氧血症)、肺无法扩张(肺复张失败)、心率过缓(心动过缓)、心脏骤停甚至死亡。 截至2026年5月6日,本次事件尚未报告任何伤害或死亡案例。 召回措施: 在Draeger公司完成呼吸机电机组件更换前,使用该设备必须进行密切、持续的监护。 Draeger公司于2026年5月7日向所有涉事客户发函,提出以下操作要求: 在呼吸机电机组件更换完成前,客户仅可在专人全程监护下继续使用设备。 若发生机械通气故障: 根据患者需求,切换至手动/自主(Man/Spont)通气模式,手动为患者通气。持续监测患者状况,重点关注氧合状态——即使短暂停止通气也可能造成伤害。切换至手动/自主通气模式后,可根据需求将“呼吸机错误!!! ”的报警优先级降级为“报警重置”。 务必确保所有Draeger公司Atlan A350、Atlan A350XL麻醉工作站的使用者,以及机构内所有负责该设备管理的人员知晓本通知。 若产品已转售/转移给第三方,需将本通知转发给接收方。 请联系Draeger公司,完成呼吸机电机组件更换,或拨打Draeger公司的服务技术支持部门电话1-800-641-2049。 请查看此网页以获取最新信息。FDA目前正在审查有关这一潜在高风险器械问题的信息,并将随着重要新信息的出现,随时向公众通报。 (美国FDA网站)
美国FDA发布关于Arrow International公司召回含有Merit Medical可撕开导管鞘透析导管套件的警示信息
发布日期:2026年5月26日 此次召回涉及将某些设备从其使用或销售地点移除。FDA已将此次召回列为最严重的类型。如果继续使用该设备,可能会造成严重伤害或死亡。受影响的产品以及对设备的处理建议如下,未发生变化。 召回产品:
FDA获悉,箭牌国际公司(Arrow International)已向受影响客户发出信函,建议将含有麦瑞通(Merit Medical)16F可撕开导管鞘的透析导管套件从使用或销售地点撤下。 受影响产品包括: 受影响产品完整清单详见:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/dialysis-catheter-kit-recall-arrow-international-removes-dialysis-catheter-kits-containing-merit 器械用途:16F可撕开导管鞘用于多个箭牌血液透析套件,包装在产品的无菌屏障内。该导管鞘用于将导管送入血管系统。 召回原因: 箭牌国际对受麦瑞通16F可撕开导管鞘召回影响的透析导管套件,启动了紧急医疗器械召回。 麦瑞通因设计缺陷召回其 16F可撕开导管鞘,该缺陷可能导致鞘管无法按预期分离。该鞘管用于多个箭牌血液透析套件中。若鞘管无法按预期分离,可能导致出血、异物、手术延迟、栓塞/血栓、导管功能受损以及血管通路的丧失。 截至2月23日,麦瑞通报告2例与该问题相关的严重伤害事件,无死亡事件。截至4月21日,箭牌国际未报告任何与该问题相关的患者伤害或死亡事件。 采取措施: 不要在箭牌国际的套件和组合中使用麦瑞通 16F可撕开导管鞘。 4月,箭牌国际公司向所有受影响的客户发送信函,建议采取以下行动: (美国FDA网站)
|



|