

|
医疗器械警戒快讯 2026年第6期(总第232期)
发布日期:2026-07-03
医疗器械警戒快讯
(总第232期) 内容提要
美国FDA发布关于Draeger公司麻醉机通气故障问题相关的警示信息 发布日期:2026年6月15日 本次召回涉及对产品进行纠正,不涉及将其从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果在未进行纠正的情况下继续使用该产品,可能会导致严重伤害或死亡。下文所列受影响产品及相关处置建议目前未发生变更。 受影响的产品: FDA获悉,Draeger已向受影响客户发出信函,建议在继续使用前对相关麻醉机进行纠正。受影响的产品如下:
受影响产品完整列表详见FDA网站:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/anesthesia-machine-correction-draeger-inc-issues-correction-atlan-a350-and-a350xl-anesthesia 产品用途: Atlan A350和A350 XL麻醉工作站适用于对成人、儿童和新生儿实施麻醉,可用于自动通气和手动通气、压力支持下的自主呼吸,以及自主呼吸。该产品用于外科手术或诊断性操作期间的吸入式麻醉。 纠正原因: Draeger公司正在扩大其2024年10月纠正的范围,将更多Atlan A350和A350 XL麻醉工作站纳入其中。本次纠正源于一项制造错误,该错误可能导致活塞式呼吸机在使用前出现故障,或在使用过程中机械通气失败。 若该错误在使用前发生(例如在待机或系统测试模式下),则机械通气无法启动。若该错误在手术过程中发生,则机械通气可能失败,产品将通过显示“Ventilator error!!!”向用户发出警报。在上述两种情况下,手动通气或自主呼吸仍然可行。为了防止伤害可能需要为患者实施手动通气。尽管通气故障报警持续存在,Atlan产品仍可提供Man/Spont通气模式、新鲜气体与麻醉剂输送以及所有监测功能,这些功能并不会受到限制。 Draeger公司将为受影响产品更换呼吸机马达组件。当地Draeger服务代表将与客户联系,安排呼吸机马达组件的更换日期。 使用受影响产品可能导致严重不良健康后果,包括缺氧(低氧血症)、肺膨胀能力丧失(无法实现肺复张)、心率减慢(心动过缓)、心脏骤停(心搏骤停)和死亡。 截至2026年5月6日,尚无受伤报告,也无死亡报告。 采取措施: 在Draeger公司完成呼吸机马达组件更换之前,应在持续严密监护下使用本产品。 5月7日,Draeger公司向所有受影响客户发出函件,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Abiomed公司自动化Impella控制器硬件问题相关的警示信息 发布日期:2026年6月11日 本次召回涉及对产品进行纠正,不涉及将其从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果在未进行纠正的情况下继续使用该产品,可能会导致严重伤害或死亡。 受影响的产品: FDA获悉Abiomed公司已向受影响客户发出信函,建议在继续使用特定自动化Impella控制器之前对其进行纠正。受影响的产品如下:
受影响产品完整列表详见FDA网站:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/heart-pump-correction-abiomed-issues-correction-automated-impella-controllers 产品用途: 自动化Impella控制器是Impella导管的主要用户控制界面。它控制Impella导管的性能,监测导管的报警状态,并提供关于导管跨主动脉瓣位置的实时位置信息。 产品更新原因: Abiomed在对服务记录进行回顾性审查时识别出部分 AIC单元(自动化Impella控制器)需要进行特定的硬件更新以解决潜在的安全问题。这些硬件更新旨在降低可能导致血流动力学支持延迟或丧失的风险,进而可能引发严重伤害或死亡。
上述所有四个问题均可能导致控制器无法启动或血流动力学支持突然中断。在所有情况下,控制台都必须更换为替代产品,而此更换过程将导致治疗受到额外延误。血流动力学支持的延迟或丧失可能因所支持患者的脆弱程度和血流动力学依赖程度而产生不同后果,并可能导致严重伤害或死亡。 截至4月14日,Abiomed公司尚未报告与此问题相关的任何严重伤害或死亡。 采取措施: 请配合Abiomed公司对受影响产品进行纠正。医院库存可继续使用。 4月20日,Abiomed公司向所有受影响客户发出函件,建议采取以下措施:
(美国FDA网站)
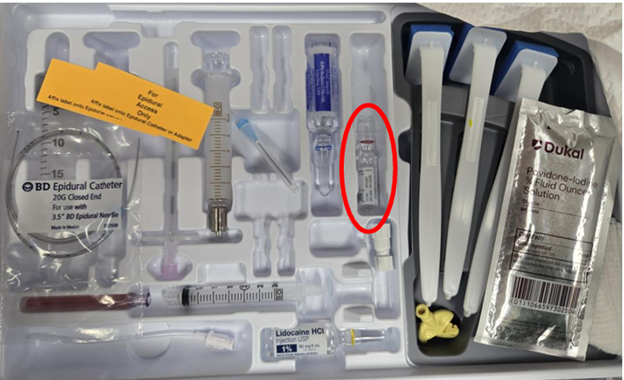
美国FDA发布关于Becton Dickinson公司含布比卡因安瓿的麻醉套件相关的警示信息 发布日期:2026年6月3日 本次召回涉及对产品进行纠正,不涉及将其从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果在未进行纠正的情况下继续使用该产品,可能会导致严重伤害或死亡。 受影响的产品:
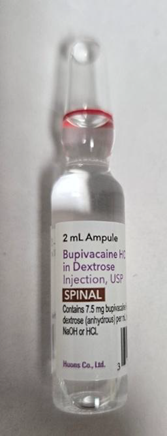
布比卡因安瓿
图中红圈标示为所涉布比卡因安瓿 FDA获悉Becton Dickinson公司已向受影响客户发出信函,建议在套件继续使用前,通过移除其中的布比卡因安瓿对全部麻醉套件进行纠正。受影响产品包括: 受影响产品完整列表详见FDA网站:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/anesthesia-kit-correction-becton-dickinson-issues-correction-bd-spinal-trays-containing-bupivacaine 产品用途: BD区域麻醉套件供专业麻醉科医师用于实施腰麻和/或硬膜外麻醉,或神经阻滞麻醉操作。 纠正原因: Becton Dickinson公司说明本次涉及的布比卡因安瓿由Huons Co., Ltd.公司生产,该公司因近期FDA检查中发现质量问题,并收到若干数量的药效不足报告,对相关安瓿实施了召回。这些安瓿被用于配置BD腰麻套件中。使用质量受损的注射药品的风险包括:局部感染、炎症反应,或麻醉效果降低。 截至4月27日,Becton Dickinson尚无报告与此问题相关的严重伤害或死亡。 采取措施: 不得使用受影响套件中的布比卡因安瓿,套件内其余组件可继续使用。 4月27日,Beckton Dickinson公司向所有受影响客户发出函件,建议采取以下措施: (美国FDA网站)
澳大利亚TGA发布关于Mindray Medical公司非充电式公共自动体外除颤器的警示信息 发布日期:2026年6月2日 Mindray Medical澳大利亚公司正在对BeneHeart D1自动体外除颤器(AED)进行产品更正,操作手册正在更新,以帮助用户更好地理解切换患者类型的操作流程。 受影响的产品:
警示原因: 当设备设置为儿童模式但用于成人患者时,存在除颤无效的风险。由于成人通常需要比儿童多得多的能量,能量不足可能难以产生足够电流使心肌完全除极。 为降低此风险,设备默认切换至成人模式,并伴有语音提示和界面图像,指示当前患者类型的状态。 尚未收到与此问题相关的伤害报告。 采取措施: 为帮助用户更好地了解BeneHeart D1自动体外除颤器(AED)切换患者类型的操作流程,Mindray Medical公司已更新操作手册,补充了更完善的指导。更新详情如下: 1.操作手册第5.4节“除颤器操作流程”中补充了以下内容:“检查除颤器上的患者类型,如果与当前患者不符,请选择下方图像以正确设置患者类型。”
患者类型切换图标 2.操作手册第5.4节“注意事项”中补充了以下内容:“当MR60或MR61电极片连接到除颤器和患者后,除颤器将切换至成人模式。若显示的患者类型与实际患者不符,需手动更改患者类型。” 此次操作手册修订仅添加补充信息,未对产品本身及其操作流程进行任何修改。 用户可查阅Mindray Medical澳大利亚公司提供的最新电子版操作手册,按照说明使用产品。 (澳大利亚TGA网站)
美国FDA发布关于KayserBetten公司KayserBett IDA系列儿科护理床相关的警示信息 发布日期:2026年5月29日 本次召回涉及对产品进行纠正,不涉及将其从使用单位或销售机构移除。FDA已经确认此次召回为最严重的等级。如果在未进行纠正的情况下继续使用该产品,可能会导致严重伤害或死亡。 受影响的产品: 产品名称:KayserBett IDA儿科护理床 FDA获悉KayserBetten公司已向受影响客户发出信函,建议所有KayserBett IDA儿科护理床在继续使用前进行纠正。 产品用途: KayserBett IDA是一种儿科护理床,旨在为居家和机构护理中患有残疾或慢性健康状况的儿童提供安全的睡眠和护理环境。该产品可调节睡眠平台高度和倾斜度,可锁定侧围栏,并具有用于调节功能的用户操作手控装置。 纠正原因: KayserBetten公司宣布其在美国销售的KayserBett IDA儿科护理床与其在美国境外销售的一款曾发生严重事故并导致儿童死亡的其他型号产品具有相似的伤害风险。 用户手册指示,当患者(儿童)无人看管时,应使用随附的钥匙锁定手控装置的调节功能。如果患者无人看管时未使用随附钥匙锁定手控装置的调节功能,患者或其他儿童可能会操作手控装置,增加将自己或他人困在床架下方或床架与地面之间空间的风险,这可能导致严重伤害或死亡。 因此,KayserBetten公司正为美国境内所有受影响的KayserBett IDA儿科护理床更换新的带有自动锁定功能的改良型手控装置。新手控装置的调节功能在每次使用后会自动锁定。授权服务技术人员将免费为客户提供手控装置更换服务。美国经销商Mobility Unlimited公司(KayserBetten美国公司)将联系客户安排更换。 截至4月22日,KayserBetten公司没有报告与该问题相关的严重伤害或死亡事件。 采取措施: 请遵循提供的临时安全措施,直至受影响产品的手控装置完成更换。 4月2日,KayserBetten公司向所有受影响客户发送信函,推荐采取以下行动: (美国FDA网站)
|

|